
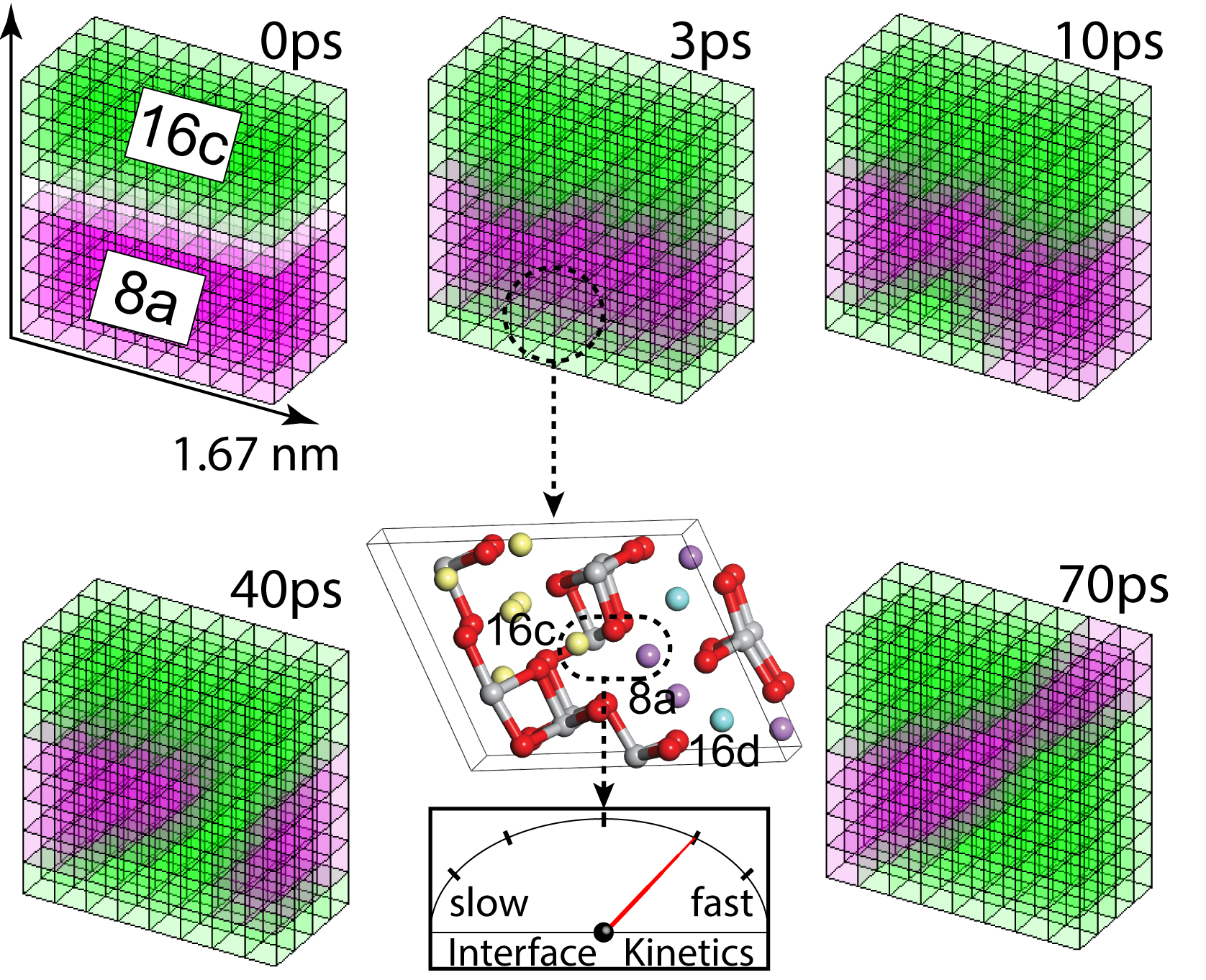
In our recent paper we computationally explored the atomic scale mechanism that governs the (dis)charging (lithiation and delithiation) of the defective spinel Li4+xTi5O12 structure, a state of the art anode material for Li-ion batteries. Although well studied and widely used commercially, the nature of the phase transition of Li4+xTi5O12 and the role of interfaces raises many questions, fed by the difficulty to distinguish the two end-member phases (Li8a and Li16c domains) with various experimental techniques.
Based on Density Functional Theory (DFT) both formation energy and Molecular Dynamics (MD) calculations were performed on Li4+xTi5O12 to explore the thermodynamics and kinetics on the atomic scale. With these calculations we showed that the interface between the two end-member phases is stabilised by the Li atoms replacing 17% of the Ti atoms in the Li4+xTi5O12 structure creating intimate mixing of the Li8a and Li16c domains. Therefore the phase transformation is a true thermodynamic first-order phase transition as the material remains phase separated, where the phase separation on a sub-nanometer scale explains both the previous solid solution interpretations and the observation of domain structures. The Li atoms replacing 17% of the Ti atoms also play a pivotal role in the kinetics of the system allowing facile transitions between a few stable local interface configurations. These transitions induce rapidly moving interfaces between the Li8a and Li16c domains, explaining the very fast response on non-equilibrium conditions such as those occurring in Li-ion batteries. Thereby, unlike in other known first-order electrode materials, the low energy associated with the coexisting phases, makes that the phase separation in LTO appears to occur spontaneously towards sub-nanometer domains.
Phase transitions play a crucial role in Li-ion battery electrodes being decisive for both the power density and cycle life. However, in-depth understanding regarding the nature of the transitions and the mechanics of the kinetics remain unexplored. This study offers a unique effort to follow the kinetic evolution of interfaces in real time using Molecular Dynamics simulations enhancing the fundamental understanding behind phase coexistence and kinetic behaviour which is relevant for improving battery performance.