
With the end of the fossil fuel era and the transition to sustainable power, energy storage mechanisms are requested to meet the new demands for energy storage of intermittent sources and high demands for electronic transportation. To come up with new solutions traditional boundaries must be challenged. Therefore professors and students from Chemical Engineering cooperate with their colleagues from Applied Physics and Material Engineering to create interdisciplinary playground, focusing on chemical energy storage we explore the limits of state of the art materials, while at same time develop new promising candidates.
Welcome
Phase-field modelling shows the limiting processes in anatase TiO2 electrodes
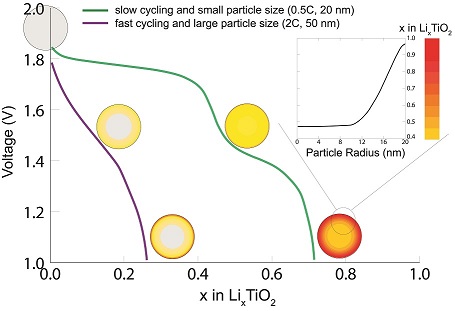 The improvement of Li-ion battery performance requires development of models that capture the essential physics and chemistry that is happening in Li-ion battery electrode materials. Phase-field modeling has shown to have this ability, providing the opportunity to gain understanding of the complex interactions happening in a battery during (dis)charge.
The improvement of Li-ion battery performance requires development of models that capture the essential physics and chemistry that is happening in Li-ion battery electrode materials. Phase-field modeling has shown to have this ability, providing the opportunity to gain understanding of the complex interactions happening in a battery during (dis)charge.
In this paper, a novel electrochemical phase-field model is presented that captures the thermodynamic and kinetic properties of lithium insertion in TiO2-anatase, a well-known and intensively studied Li-ion battery electrode material. Previous elaborate experimental work on anatase TiO2 provides all parameters necessary for the phase-field simulations, giving the opportunity to gain fundamental insight and validate this phase-field model.
The model shows that, unlike other electrode materials, the kinetic limitations of individual anatase particles limit the performance of full electrodes. Hence, improving the performance of anatase TiO2 electrodes requires increasing the Li-diffusion in anatase particles.
DOI: 10.1103/PhysRevMaterials.1.025404
Thermodynamics and Kinetics of Na-ion Insertion into Ti oxide Structres
 First principle DFT calculations are used to study the thermodynamic and kinetic properties of Na-ion insertion in TiO2 hollandite, a potential anode for Na-ion batteries. The experimentally observed phase transformation from tetragonal TiO2 (I4/m) to monoclinic Na0.25TiO2 (I2/m) is confirmed. At high Na-ion concentrations the calculated formation energies predict a first order phase transition towards the layered O’3-Na0.68TiO2 structure. Further sodiation initiates a solid solution reaction towards the layered O3-NaTiO2 phase, which was recently brought forward as a promising anode for Na-ion batteries. This transformation irreversibly transforms the one dimensional Hollandite tunnel structure into the layered structure, and potentially brings forward an alternative route towards the preparation of the hard to prepare O3-NaTiO2 material. Energy barrier calculations reveal fast Na-ion diffusion at low concentrations and sluggish diffusion upon reaching the N0.25TiO2 phase, rationalizing why experimentally the Na0.5TiO2 phase is not achieved. Detailed analysis of the kinetic behaviour in the hollandite structure via Molecular Dynamics simulations reveal the importance of correlated atomic motions and dynamic lattice deformations for the Na-ion diffusion. In addition, exceptional Na-ion kinetics were predicted for the layered O’3-Na0.75TiO2 phase through a dominating divacancy hopping mechanism.
First principle DFT calculations are used to study the thermodynamic and kinetic properties of Na-ion insertion in TiO2 hollandite, a potential anode for Na-ion batteries. The experimentally observed phase transformation from tetragonal TiO2 (I4/m) to monoclinic Na0.25TiO2 (I2/m) is confirmed. At high Na-ion concentrations the calculated formation energies predict a first order phase transition towards the layered O’3-Na0.68TiO2 structure. Further sodiation initiates a solid solution reaction towards the layered O3-NaTiO2 phase, which was recently brought forward as a promising anode for Na-ion batteries. This transformation irreversibly transforms the one dimensional Hollandite tunnel structure into the layered structure, and potentially brings forward an alternative route towards the preparation of the hard to prepare O3-NaTiO2 material. Energy barrier calculations reveal fast Na-ion diffusion at low concentrations and sluggish diffusion upon reaching the N0.25TiO2 phase, rationalizing why experimentally the Na0.5TiO2 phase is not achieved. Detailed analysis of the kinetic behaviour in the hollandite structure via Molecular Dynamics simulations reveal the importance of correlated atomic motions and dynamic lattice deformations for the Na-ion diffusion. In addition, exceptional Na-ion kinetics were predicted for the layered O’3-Na0.75TiO2 phase through a dominating divacancy hopping mechanism.DOI: 10.1021/acs.chemmater.6b03928
The fine line between a two-phase and solid-solution phase transformation and highly mobile phase interfaces in spinel Li4+xTi5O12
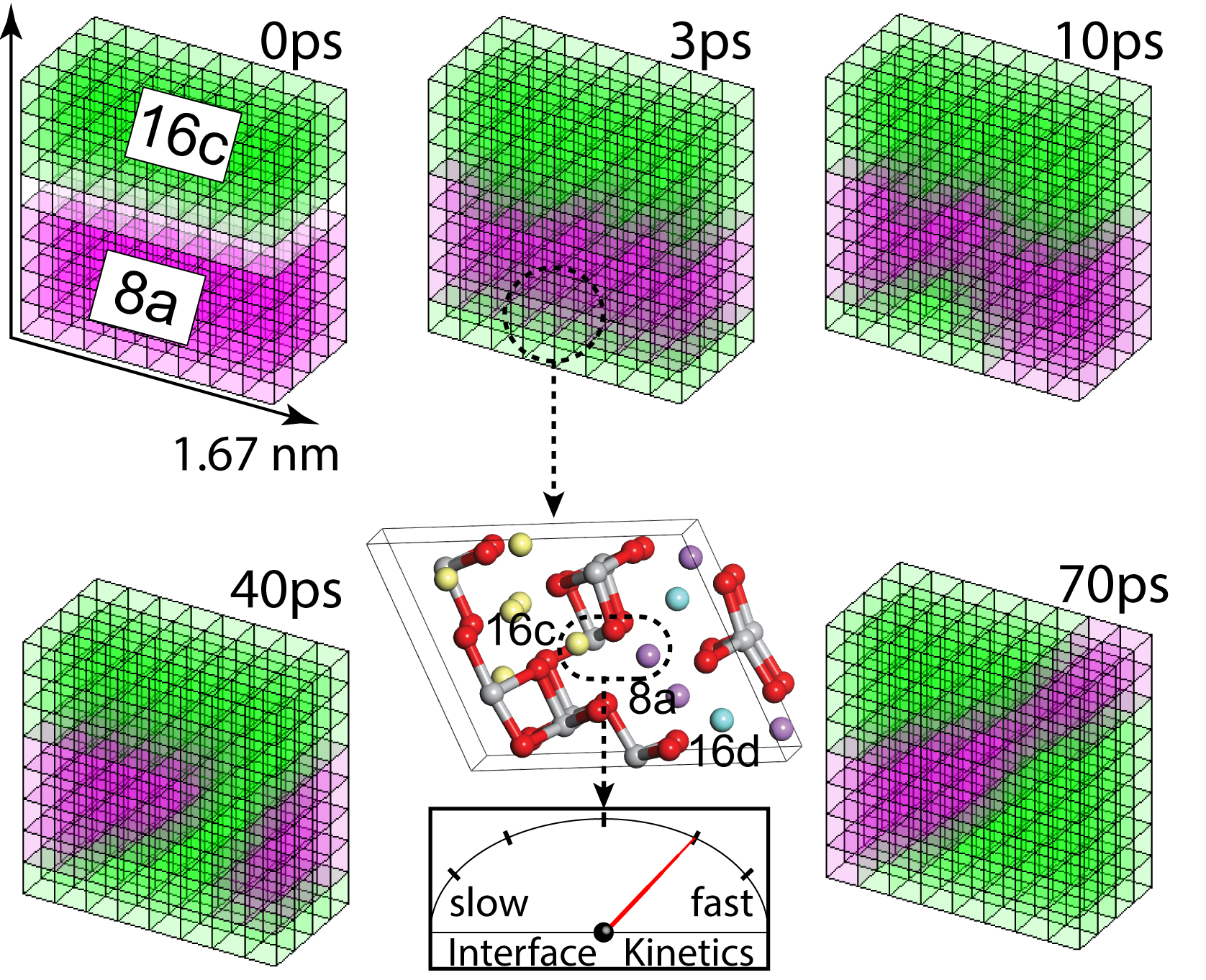
In our recent paper we computationally explored the atomic scale mechanism that governs the (dis)charging (lithiation and delithiation) of the defective spinel Li4+xTi5O12 structure, a state of the art anode material for Li-ion batteries. Although well studied and widely used commercially, the nature of the phase transition of Li4+xTi5O12 and the role of interfaces raises many questions, fed by the difficulty to distinguish the two end-member phases (Li8a and Li16c domains) with various experimental techniques.
Based on Density Functional Theory (DFT) both formation energy and Molecular Dynamics (MD) calculations were performed on Li4+xTi5O12 to explore the thermodynamics and kinetics on the atomic scale. With these calculations we showed that the interface between the two end-member phases is stabilised by the Li atoms replacing 17% of the Ti atoms in the Li4+xTi5O12 structure creating intimate mixing of the Li8a and Li16c domains. Therefore the phase transformation is a true thermodynamic first-order phase transition as the material remains phase separated, where the phase separation on a sub-nanometer scale explains both the previous solid solution interpretations and the observation of domain structures. The Li atoms replacing 17% of the Ti atoms also play a pivotal role in the kinetics of the system allowing facile transitions between a few stable local interface configurations. These transitions induce rapidly moving interfaces between the Li8a and Li16c domains, explaining the very fast response on non-equilibrium conditions such as those occurring in Li-ion batteries. Thereby, unlike in other known first-order electrode materials, the low energy associated with the coexisting phases, makes that the phase separation in LTO appears to occur spontaneously towards sub-nanometer domains.
Phase transitions play a crucial role in Li-ion battery electrodes being decisive for both the power density and cycle life. However, in-depth understanding regarding the nature of the transitions and the mechanics of the kinetics remain unexplored. This study offers a unique effort to follow the kinetic evolution of interfaces in real time using Molecular Dynamics simulations enhancing the fundamental understanding behind phase coexistence and kinetic behaviour which is relevant for improving battery performance.
The Significance of Elemental Sulfur Dissolution in Li-S Batteries
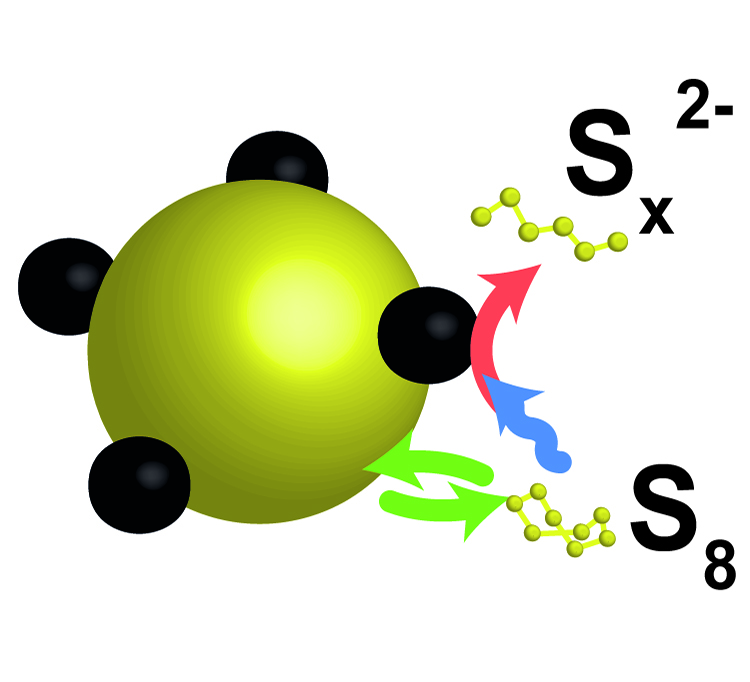 In an effort to keep up with the increasing demands for energy storage, lithium sulfur (Li-S) batteries receive a lot of research attention as they are considered one of the most promising technologies to replace the current lithium-ion batteries. Sulfur has the ability to reversibly store 2 electrons per atom and is an abundant, benign, and low cost material. However, sulfur also has an extremely low electrical conductivity. Using a highly resistive active material in a battery normally means severe (ohmic) losses, and is therefore seen as a large obstacle for the efficient storage of energy. This is in conflict with the relative ease with which sulfur can be applied as electrode material, evidenced by the many promising results obtained for Li-S batteries over the last few years.
In an effort to keep up with the increasing demands for energy storage, lithium sulfur (Li-S) batteries receive a lot of research attention as they are considered one of the most promising technologies to replace the current lithium-ion batteries. Sulfur has the ability to reversibly store 2 electrons per atom and is an abundant, benign, and low cost material. However, sulfur also has an extremely low electrical conductivity. Using a highly resistive active material in a battery normally means severe (ohmic) losses, and is therefore seen as a large obstacle for the efficient storage of energy. This is in conflict with the relative ease with which sulfur can be applied as electrode material, evidenced by the many promising results obtained for Li-S batteries over the last few years.
In our recent publication, we assessed the implications of the non-conductive characteristics of sulfur for the operating principles of the Li-S battery. By applying a simple but effective battery cell we showed that the dissolution of elemental sulfur, despite its low solubility in the organic solvent of the electrolyte, plays a significant role in the operation mechanism of the Li-S battery. From the results it was evident that sulfur can participate in the redox reactions as a dissolved molecule. In doing so the low conductivity of bulk sulfur is effectively circumvented; the electrons do not need to go through the bulk sulfur, but the dissolved sulfur goes to the electrons. Based on the electrochemical response of the special cells the researchers furthermore indicate that the non-conducting solid sulfur can be utilized indirectly by interactions with intermediate products that are formed during battery operation. These factors explain the relative ease with which this insulator can be applied as electrode material in liquid electrolyte cells.
Increasing Li-ion conductivity in argyrodite solid electrolytes
 The use of liquid electrolytes makes current batteries prone to dangerous thermal runaway reactions, igniting the battery, as seen recently in the Samsung Note 7 phones. To make batteries safer the liquid electrolyte can be replaced by a solid electrolyte, which is not flammable. However, before solid electrolytes can be used the Li-ion conductivity must be equally good as in liquid electrolytes.
The use of liquid electrolytes makes current batteries prone to dangerous thermal runaway reactions, igniting the battery, as seen recently in the Samsung Note 7 phones. To make batteries safer the liquid electrolyte can be replaced by a solid electrolyte, which is not flammable. However, before solid electrolytes can be used the Li-ion conductivity must be equally good as in liquid electrolytes.
Currently several solid electrolytes are known with Li-ion conductivities comparable to that in liquid electrolytes, one of which are lithium argyrodites. To gain a better understanding of Li-diffusion in lithium argyrodites density functional theory molecular dynamics simulations have been performed.The simulations showed that the position of halogen atoms in the crystals has a large impact on the Li-ion conductivity, and that altering the halogen positions in lithium argyrodites during synthesis could increase the Li-ion conductivity of these materials.
The simulation results show that the Li-ion conductivity in these materials can a factor of 2 larger than the currently highest value. (Article)